1.7 Disruption of chromatin architecture in disease
The insulating function of TADs, which restrict promoter-enhancer interactions (Symmons et al. 2014), suggests that alterations of TAD structures may induce ectopic interactions between regulatory elements and genes in neighboring TADs, leading to gene dysregulation (Fig. 1.4). Consistently, genetic manipulation of specific TAD boundaries can change the surrounding interaction patterns and thus affect the expression of nearby genes. After experimental deletion of a 58-kb region encompassing a TAD boundary in mouse ESC, interactions between adjacent TADs increased significantly and genes in neighboring TADs were upregulated (Nora et al. 2012). More precise deletion or inversion of CTCF binding site at loop anchors in TAD boundaries altered local chromatin architecture and nearby gene expression (Dowen et al. 2014; Guo et al. 2015; Narendra et al. 2015).
The alteration of chromatin contacts upon experimental disruptions of TAD boundaries leads to the question whether disruption of TADs could also be and mechanism of disease pathology. This hypothesis was initially discussed by studying the etiology of the Liebenberg syndrome, in which an upper-limb malformation phenotype is caused by a deletion in the vicinity of the PITX1 gene (Spielmann and Mundlos 2013). PITX1 becomes thereby accessible to an enhancer with specific activity in lower-limb and was miss-expressed in a corresponding mouse model (Spielmann et al. 2012). Overlap of this deletion with later identified TAD boundaries leads to the hypothesis of a regulatory effect mechanism of boundary deletions referred to as enhancer adoption.
An association of TAD disruptions with diseases was first shown by computational analysis integrating TAD structures, with tissue-specific enhancers, and large chromosomal deletions, which were associated with clinical phenotypes (Ibn-Salem et al. 2014). In this study, the Human Phenotype Ontology database (Köhler et al. 2014) was used to relate the phenotypes of 922 deletion cases from the DECIPHER database (Firth et al. 2009) to monogenic diseases associated with genes in or adjacent to the deletions. This was used to differentiate for each deletion case between two possible pathogenic effect mechanism that best explains the phenotypes observed in the patients (Fig. 1.4). Many deletions were best explained by a gene dosage mechanism and haploinsufficiency of genes located within the deletion. However, about 12 percent of cases were best explained by a TAD boundary disruption and specific combinations of tissue-specific enhancers and genes in adjacent TADs. Importantly, randomization of phenotype data showed that this enhancer adaption mechanism was significantly more frequent than expected by chance (Ibn-Salem et al. 2014). Therefore, this study shows for the first time an association of TAD disruption with genetic diseases and describes a regulatory effect mechanism of TAD disruptions that need to be further investigated and has to be considered when interpreting genetic variations in patient genomes.
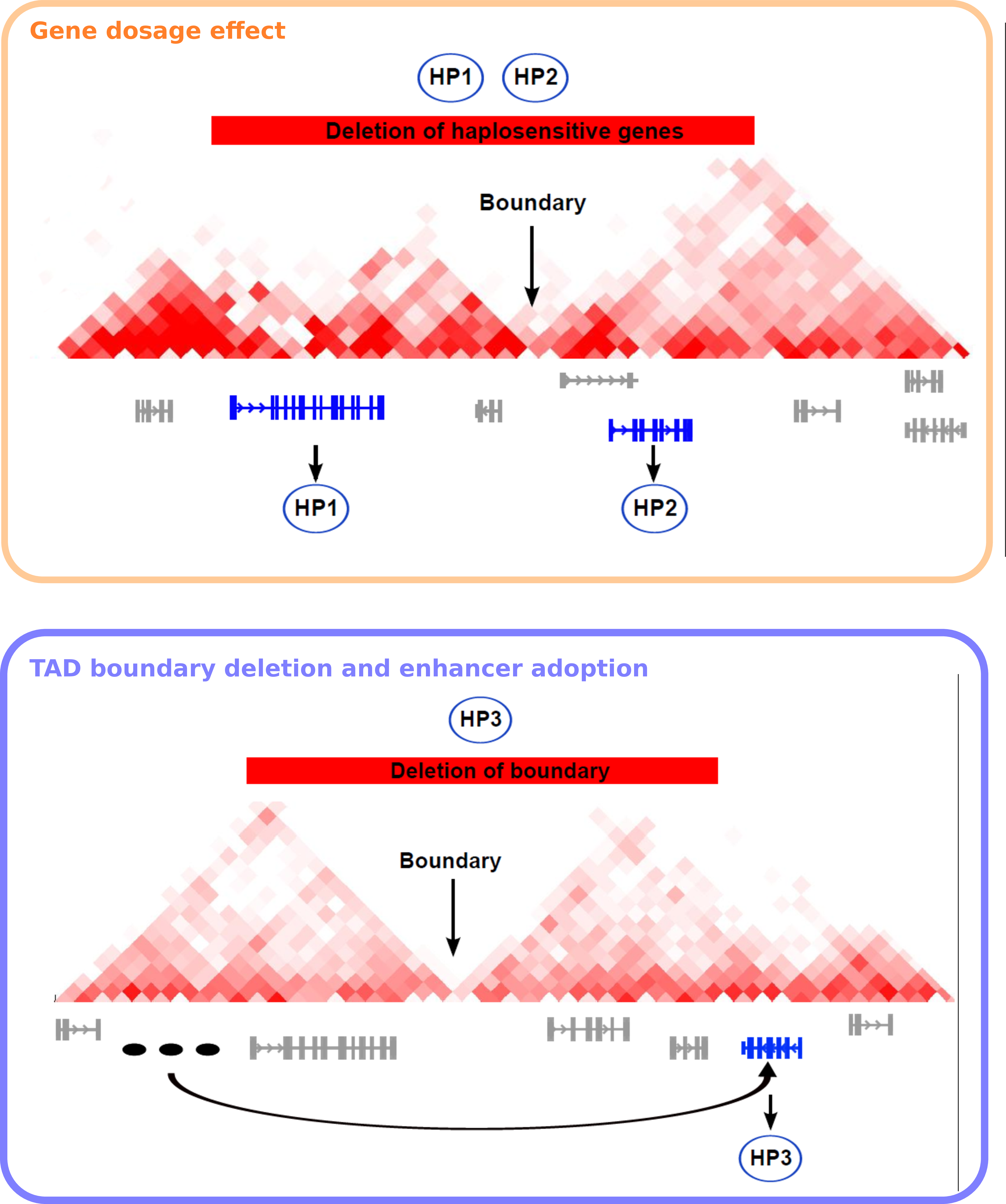
Figure 1.4: Models of pathomechanisms by chromosomal deletions involving TAD disruption In each panel, an exemplary deletion is shown as a red bar, a TDB is indicated with a black arrow, and genes associated with the phenotypes of the CNV patient are shown in blue, other genes in gray. Phenotypic abnormalities are represented as exemplary HPO terms (HP1, HP2, and HP3). Three tissue-specific enhancers are shown in (B) as black ovals. (A) Gene-dosage effect. A deletion leads to a reduction in the dosage of haplosensitive genes located within the CNV. The individual with the deletion has two phenotypic abnormalities (HP1, HP2) resulting from deletion of two haplosensitive genes. A Mendelian disease related to mutations in the first gene is associated with HP1, and a Mendelian disease related to mutations in the second gene is associated with HP2. (B) TAD boundary deletion and enhancer adoption. Removal of the topological domain boundary allows the tissue-specific enhancer inappropriately to activate a phenotypically relevant gene located adjacent to the deletion, a phenomenon that we refer to as enhancer adoption. In this case, the individual with the deletion has a phenotypic abnormality (HP3) that is also seen in individuals with a Mendelian disease related to a mutation in the gene adjacent to the deletion. Figure adapted from (Ibn-Salem et al. 2014).
Interestingly, these initial results are largely confirmed by more recent studies investigating structural variants that disrupt TADs and lead to ectopic gene expression in genetic diseases (Lupiáñez et al. 2015; Franke et al. 2016; Redin et al. 2017) and cancers (Northcott et al. 2014; Hnisz et al. 2016b; Weischenfeldt et al. 2016). This is discussed in more detail below (Chapter 6).
Together, these observations lead to the questions whether other types of structural variants, like translocations and inversions, could also disrupt TADs and regulatory interactions of genes. To analyze such a position effect, we need careful integration of diverse genomic and phenotypic data. Computational tools could predict the impact of rearrangement breakpoints on TAD disruption and potential regulatory effects on phenotypically relevant genes. Such a method would improve molecular diagnoses of many patients with variants of unknown significance.