6.4 Gene expression changes by altered TADs in disease
The functional importance of TADs is supported by their stability during evolution. However, if TADs are indeed crucial for proper gene regulation during development, one would expect that TAD disruptions are associated with disease and lead to phenotypes by dysregulation of phenotypically relevant genes.
We analyzed balanced chromosomal rearrangements in 17 subjects with diverse pathogenic phenotypes (Chapter 4). The rearrangement breakpoints are in non-coding regions but frequently disrupt TADs. Furthermore, we reported disruption of long-range chromatin interactions between several enhancers and genes whose annotated clinical features are strongly associated with the subjects phenotypes. For some candidate genes, we confirm gene expression changes in cell lines derived from subjects with such rearrangements. These detrimental effects of rearrangements in disease genomes are consistent with our comparative genomic analysis, finding selective pressure on TAD disruptions.
The causal relationship between TAD disruption, miss-expression and pathogenic phenotypes has been studied recently in more detail (reviewed for example in Krijger and Laat (2016); Achinger-Kawecka et al. (2016); Yu and Ren (2017); Andrey and Mundlos (2017)). In these studies, the authors linked observed pathogenic phenotypes of congenital diseases and cancers to structural variations identified in the subjects genomes. The observed phenotypes could not be explained, when only the disruption of protein-coding sequence or gene dosage was considered. However, when considering the genome folding structure and TADs together with enhancer activity, the pathogenic effect mechanism became apparent.
Deletions of TAD boundaries can lead to ectopic activation of genes by enhancers in neighboring TADs (Fig. 6.3A). This effect mechanism was first shown to be relevant by computationally integrating hundreds of deletions with tissue-specific enhancers, phenotype-gene associations, and TAD positions (Ibn-Salem et al. 2014). Such an enhancer adaption mechanism could best explain a significant proportion of deletion cases.
The development of CRISPR-derived methodologies allows validating miss-expression due to ectopic contacts by experimentally induced structural variants in mice (Kraft et al. 2015; Andrey and Mundlos 2017). These approaches could experimentally validate that deletion of the centromeric or telomeric boundary of a TAD containing the EPHA4 gene causes activation of IHH or PAX, leading to brachydactyly or polydactyly, respectively (Lupiáñez et al. 2015). Interestingly and in line with the insulating function of TAD boundaries, introducing a slightly shorter deletion, that did not overlap the TAD boundary, did not cause up-regulation (Lupiáñez et al. 2015). Other examples include the locus of the LMNB1 gene (Giorgio et al. 2015). Deletion of a TAD boundary upstream of LMNB1 result in ectopic contact of enhancers with the LMNB1 promoter, and overexpression of LMNB1. This pathomechanism leads to autosomal dominant adult-onset demyelinating leukodystrophy (Giorgio et al. 2015).
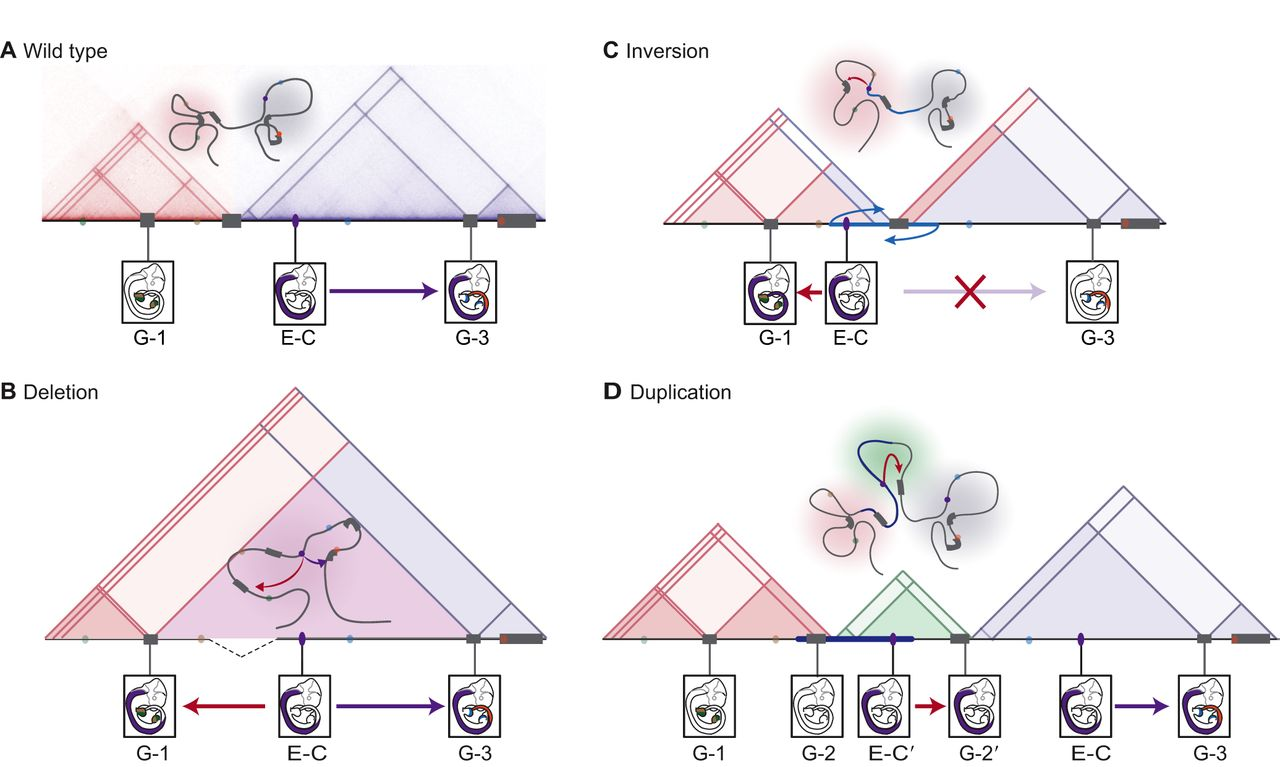
Figure 6.3: Structural variants can affect TAD structure and enhancer-promoter interactions. Structural variants can induce ectopic increase or loss of gene expression leading to disease. Here, topologically associating domains (TADs) are represented by shaded triangles, genes by gray blocks, and enhancers by colored ovals. Gene expression patterns are driven by enhancers in E10.5 mouse embryos are shown. A representative chromatin configuration is shown above the TADs. (A) In the wild-type chromatin conformation, Enhancer C (E-C) controls Gene 3 (G-3) in the neural tube. (B) The deletion of a TAD boundary element leads to ectopic contact between Enhancer C (E-C) and gene 1 (G-1) (red arrow), which results in the ectopic expression of Gene 1 in the neural tube. (C) In the case of a rearrangement through an inversion (blue arrows) or translocation that leads to the repositioning of functional elements, Enhancer C (E-C) from a neighboring TAD is free to activate Gene 1 (G-1), resulting in the ectopic expression of Gene 1 in the neural tube. The inversion also leaves Enhancer C insulated from its native target Gene 3 (G-3) by a boundary, resulting in the loss of Gene 3 expression in the neural tube. (D) The duplication of a region allows a new chromatin domain (a neo-TAD; green triangle) to form that contains regulatory region(s) and gene(s), which produce new expression patterns. Here, the duplicated Enhancer C′ (E-C′) and Gene 2′ (G-2′), which both locate to the insulated neo-TAD, produce the ectopic expression of Gene 2′ in the neural tube. Figure and figure caption adapted from (Andrey and Mundlos 2017).
Depending on their size and positioning, also duplications can bring enhancers into contact with gene promoters, which were before separated by a TAD boundary (Fig. 6.3D). Duplications involving no regulatory elements and contained entirely within a TAD (intra-TAD) have in general no major effect on genes in the same TAD, as enhancer-promoter contacts are mainly invariant of genomic distance within TADs (Symmons et al. 2016). This lack of regulatory phenotype can also explain why paralog genes are often created by tandem-duplications within TADs during evolution without affecting the regulatory environments of other genes (Chapter 2). However, when an intra-TAD duplication contains regulatory environments with several enhancers, increased regulatory inputs can lead to over-expression. This mechanism leads, for example, to upregulation of the SOX9 gene and, in turn, causes female to male sex reversal (Franke et al. 2016). However, further extension of the duplication at the SOX9 locus, encompassing a TAD boundary and the nearby KCN72 gene, leads to the formation of a new TAD, called neo-TAD. Since both, SOX9 enhancers and the KCN72 gene are included in the duplicated regions, KCN72 gets activated by the SOX9 enhancers leading to Cooks syndrome (Franke et al. 2016).
Beside deletions and duplications, balanced chromosomal abnormalities (BCA) like inversion or translocation can disrupt TADs and lead to similar effect mechanism without gain or loss of genetic material. Inversions of TAD boundaries can bring promoters under the control of an enhancer, that was before in different TADs (Fig. 6.3C). Such a mechanism leads for example to F-syndrome, where inversion of the centromeric boundary of the EPHA4 TAD cause activation of the WNT6 gene (Lupiáñez et al. 2015). Another recent study analyzed BCAs in 273 subjects with a spectrum of congenital abnormalities and find 7.3% of them disrupting TADs encompassing known syndromic loci. Eight rearrangement breakpoints were localized in a single TAD containing the MEF2C gene and resulted in decreased expression of MEF2C in these subjects (Redin et al. 2017).
Also, somatic mutation can disrupt TAD boundaries and lead to cancers. For example, in T-cell acute lymphoblastic leukemia, two oncogenes, TAL1 and LMO2 are close to a TAD boundary and get activated upon boundary deletion in HEK-293T cells (Hnisz et al. 2016b). Furthermore, in IDH mutant gliomas, TET proteins, which are involved in active demethylation, are repressed. This effect leads to hypermethylation of several CTCF binding sites and, in turn, decreased insulation functions of TAD boundaries. This result in ectopic contact of constitutively active enhancers and the oncogene PDGFRA leading to its upregulation (Flavahan et al. 2016). In conjunction with these findings, CTCF and cohesin binding sites are frequently mutated by single nucleotide variants in several cancer types, suggesting oncogene activation by TAD disruptions as a common mechanism in cancers (Katainen et al. 2015; Yu and Ren 2017).
Together, these studies confirm our findings of computationally predicted pathomechanism of TAD disruptions upon deletions (Ibn-Salem et al. 2014) or balanced rearrangements (Chapter 4). TAD disruptions can lead to miss-expression not only during evolution but also in disease manifestation. Therefore, it will be increasingly important to take the three-dimensional genome folding structure into account to interpret the effect of genetic variants.