6.1 Co-regulation of functionally related genes in TADs
The ability to measure gene expression genome-wide in many different tissues and conditions allowed the observation of clusters of co-expressed genes in higher eukaryotes (Boutanaev et al. 2002; Purmann et al. 2007). It was previously speculated that the structure of the chromatin and cis-acting units might be responsible for the observed co-expression (Sproul et al. 2005; Purmann et al. 2007). The ability to measure chromatin interactions leads to the discovery and characterization of TADs and enforces the question whether TADs insulate regulatory units in the genome to allow co-regulation of functionally similar genes.
To study the interplay between TADs, gene co-regulation, and evolution, we decided to focus on pairs of paralog genes. Paralogs arise from gene duplication events during evolution. Because of their homology and resulting sequence similarity, paralog genes often encode proteins with related functions. This makes them an exceptional model for functionally related and co-regulated genes. Indeed, in gene expression data from various sources across different cell types and tissues, paralogs have significantly increased expression correlation compared to other close genes (Chapter 2).
The main challenge in statistically analyzing paralog gene pairs was their bias for short genomic distances. Most duplications appear to be created by tandem duplications in direct orientation (Newman et al. 2015), which explains the clustering of paralogs in the genome and enrichment for being transcribed from the same DNA strand. These properties complicated our analysis by the need for an adequately sampled control set of gene pairs.
However, the development of careful sampling techniques results in control gene pairs that have similar properties regarding genomic distance, transcription strand, number of enhancers per gene, and the distance of enhancers to genes. These approaches allowed us to compare features of paralog gene pairs to random expectations in a statistically robust manner.
Our results show that paralogs are significantly enriched in TADs, frequently share the same regulatory enhancer and have increased Hi-C contacts, even when they are more than 1 Mb apart in the linear genome. These results show that evolutionary and functionally related genes tend to be co-regulated within TADs. Importantly, this highlights a functional organization of the three-dimensional genome, in which domain organization segregates distinct regulatory environments (Fig. 6.1).
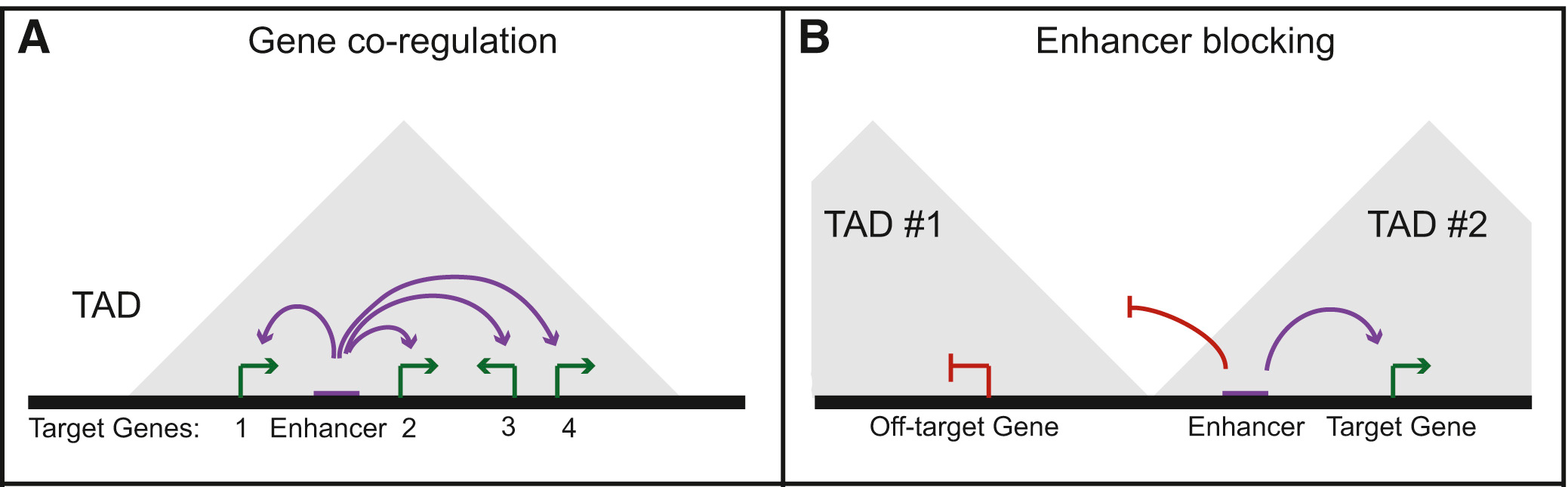
Figure 6.1: Co-regulation by shared enhancers in TADs. (A) Example diagram showing the co-regulation of multiple genes by a single regulatory element within a TAD. (B) Diagram of the potential for TAD boundaries to serve an enhancer blocking role that restricts enhancers to target genes within the same TAD. Figure adapted from (Dixon et al. 2016).
The association of gene expression with gene localizing in TADs is consistent with a very recent computational study with the aim to separate the proportion of expression associated with genome organization from independent sources. A large fraction of expression variance can be attributed to the positioning of genes in genome architecture and is highly informative for TAD activity and organization (Rennie et al. 2018).
Together, with results from many other studies (Bonev and Cavalli 2016; Andrey and Mundlos 2017; Hnisz et al. 2016a), our results support the notion of TADs as functional units of chromosomes in which related genes are co-regulated.