4.4 Discussion
Structural variation of the human genome, either inherited or arising by de novo germline or somatic mutations, can give rise to different phenotypes through several mechanisms. Chromosome rearrangements can alter gene dosage, promote gene fusions, unmask recessive alleles, or disrupt associations between genes and their regulatory elements. The traditional clinical focus of studying genes disrupted by chromosome rearrangements has shifted to also assess regions neighboring these variants.(Ordulu et al. 2016) This search for positional effects has been particularly important in the analysis of chromosome rearrangements associated with different clinical conditions and disrupting non-annotated genomic regions.(Zhang and Wolynes 2015; Spielmann and Mundlos 2016)
The study of chromatin conformation has been requisite in the analysis of such non-coding rearrangements. DNA is organized in the three-dimensional nucleus at varying hierarchical levels that are important for the regulation of gene expression,(Wit and Laat 2012) with primary roles in embryonic development and disease.(Bonev and Cavalli 2016) Several studies have analyzed the impact of structural variants in disruption of the regulatory chromatin environment leading to disease;(Lupiáñez et al. 2015; Gröschel et al. 2014; Visser et al. 2012; Roussos et al. 2014; Giorgio et al. 2015; Ibn-Salem et al. 2014) these studies have set the precedent for integrative analyses of disrupted chromatin conformation to expedite functional annotations of non-coding chromosome rearrangements.
We tested the possibility of utilizing chromatin contact information to dissect chromosome rearrangements which disrupt non-coding chromosome regions in clinical cases. We focused on 17 subjects from DGAP, 12 with available clinical microarray information, with different rare presentations and de novo non-coding BCAs classified as VUS. Of these, 15 corresponded to translocations and two were inversions. These cases represent ~11% of the total number of sequenced DGAP cases, which makes our predictions even more significant for future potential treatment or management of subjects who would not otherwise obtain a clinical diagnosis. Utilizing publicly available annotated genomic and regulatory elements, chromatin conformation capture information, predicted enhancer-promoter interactions, phenomatch scores, as well as haploinsufficiency and triplosensitivity information for all genes surrounding the BCA breakpoints at different window sizes (+-3 and +-1 Mb as well as BCA-containing TAD positions), we discovered 16 genes for 11 DGAP cases that are top-ranking position effect candidates for the subjects’ clinical phenotypes (Table 4.1).
We observed that eight of the sequenced DGAP BCA breakpoints, corresponding to six DGAP cases (DGAP017, 176, 249, 275, 288 and 322), overlapped reported annotated and predicted enhancers and DHS sites. Disruption of these regulatory elements could potentially cause improper gene expression or repression through altered enhancer-promoter interactions or interactions with other DHS-associated elements such as insulators and locus control regions, among others. In fact, four of the breakpoints that disrupt annotated DHS sites and enhancers have been shown to establish chromatin contacts with our top position effect candidate genes in the region in Hi-C data of H1-hESC cells at 40 Kb resolution (Table S18). For example, the DGAP275_B breakpoint is involved in a chromatin interaction that puts it into physical proximity with POLE and ANKLE2, DGAP288_B contacts SOX9, and DGAP176_B interacts with ACSL4. Three additional breakpoints from DGAP111, 249 and 287 overlap CTCF binding sites. CTCF binding sites are enriched in TAD boundaries,(Dixon et al. 2012) and the elimination of these binding sites could potentially induce gene expression or other functional changes through alteration of the structural regulatory landscape of the region.(Lupiáñez et al. 2015)
There are nine DGAP cases (DGAP113, 126, 138, 153, 163, 252, 315, 319 and 329), six with normal arrays and two with benign CNVs, for which no overlap with genomic or other regulatory elements was detected. These cases thus represent events in which position effects are most likely caused by alteration of the underlying chromatin structure itself. This hypothesis is supported by detection of a vast number of disrupted chromatin contacts in four different cell lines (H1-hESC, IMR90, GM06990, GM12878) at different Hi-C window resolutions, 32 breakpoints included in H1-hESC TADs,(Dixon et al. 2012) and the separation of 193 genes from one and up to 91 of their predicted enhancers after the occurrence of the BCAs (Table S14). For example, SOS1, one of the most significant candidates in explaining DGAP163’s global developmental delay, dysmorphic/distinctive facies and hearing loss, as observed in Noonan Syndrome 1 (NS1 [MIM: 163950]), is separated from its interaction with 88 predicted enhancers (Figure 4.3), and exhibited a decrease in expression in DGAP163-derived LCLs. However, NS1 is caused by autosomal dominant mutations in SOS1; we hypothesize that the reduced expression of SOS1 might affect the RAS/MAPK signaling pathway and generate clinical features not completely overlapping those of NS1; however, this possibility remains to be functionally tested and complimented with analyses of genomic single nucleotide variants. A similar approach could be explored for DGAP275, where we hypothesize that POLE, associated with the facial dysmorphism, immunodeficiency, livedo, and short stature syndrome (FILS [MIM: 615139]) in an autosomal recessive manner,(Pachlopnik Schmid et al. 2012) may contribute to the extreme short stature observed in this DGAP subject; and ZEB2, etiologic for Mowat-Wilson syndrome (MOWS [MIM: 235730]) in an autosomal dominant manner (OMIM#235730), may potentially explain the hypotonia and neurological features observed in DGAP329 but not present all of the dysmorphic features or medical/non-neurologic phenotype of MOWS. Overall, more candidate genes will need to be analyzed rigorously to assess the validity of our position effect predictions and the disruption of important chromatin regulatory elements. Nonetheless, insight into the molecular pathway of disorders may be forthcoming from our approach and of value in the management of some individuals.
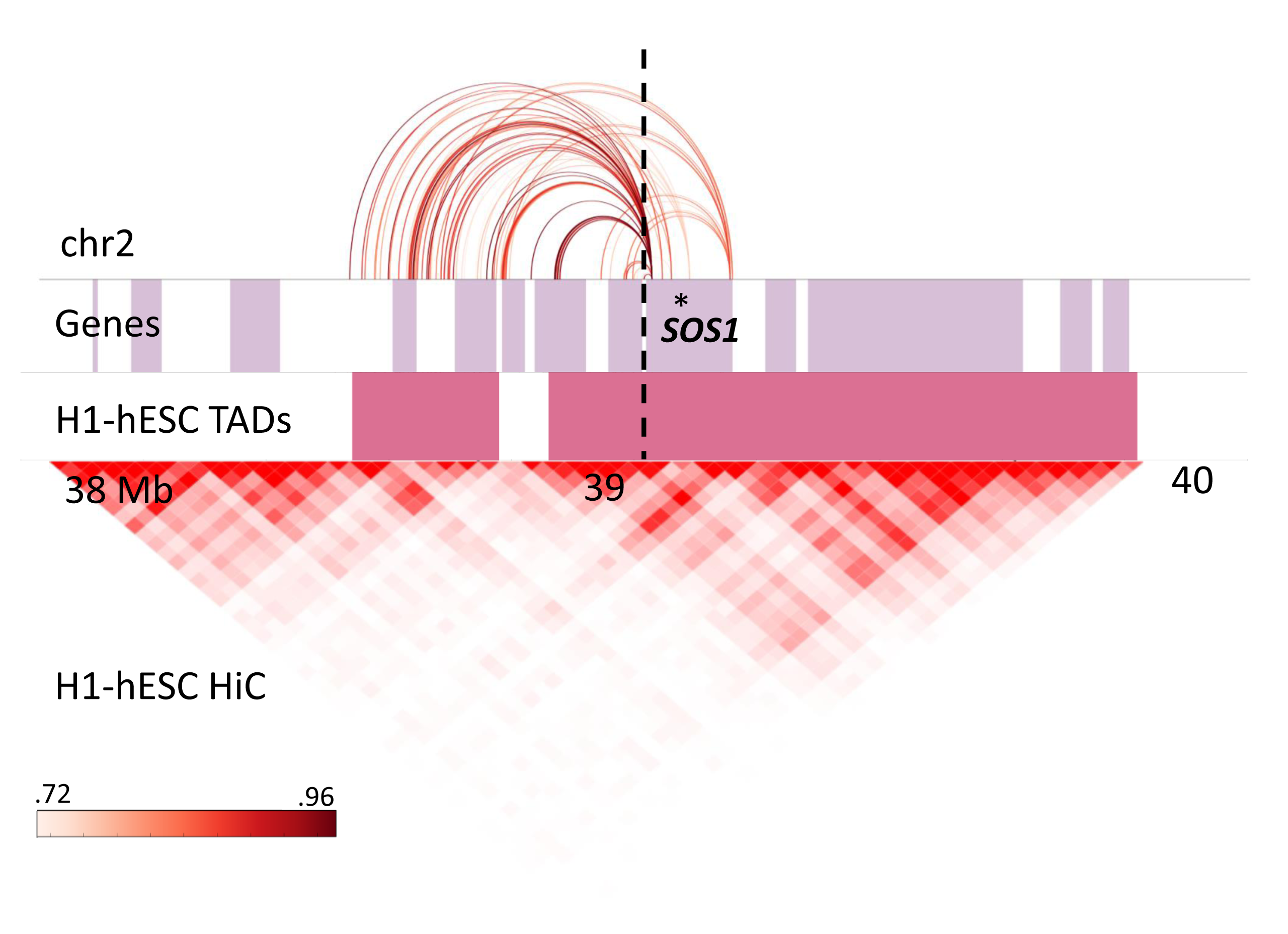
Figure 4.3: Disrupted enhancer-promoter DHS interactions predicted for SOS1 (gene position indicated by asterisk). The color graded rectangle represents the correlation values for the interactions as reported by ENCODE. The dashed line indicates the translocation breakpoint position in chromosome 2. Lilac colored rectangles represent genes, and pink rectangles show TAD positions annotated in H1-hESC.
All predicted candidate genes have different lines of evidence supporting their selection, starting with a significant phenomatch score that correlates annotated gene phenotypes to those observed in the DGAP cases. HI and triplosensitivity evidence, inclusion in TAD regions, as well as HI scores build upon this selection, and can help laboratories and clinicians focus in subsequent analyses on candidates of their interest. As of now, the “top-ranking” candidates have the highest number of evidence supporting their selection; however, there are also 102 second-tier candidates for the 17 analyzed DGAP cases within +-1 Mb analysis windows which may well play a functional role. Presently, we are unable to give “weights” to any of these selection criteria (i.e., a gene with a high phenomatch score and no evidence of HI is “more significant” than a gene with a medium phenomatch score and evidence of HI) mainly for two reasons: (i) we would need to collect more examples, which might not be easy to find and require a tremendous curation effort, and (ii) we need to understand the possibility, suggested by our results, that more than one gene may be contributory in the clinical presentation of the DGAP subjects, either acting simultaneously or throughout development. Moreover, many of the candidates have recessive inheritance modes, which make it necessary to assess the mutational status of both alleles as well as additional sequence variants not captured by our BCA breakpoint sequencing and the microarrays. Future in-depth exome, DNA and RNA sequencing as well as Hi-C experiments will provide a comprehensive view of the contribution of sequence variants, disruption of chromatin contacts, and changes in gene expression in the DGAP disease etiologies, such that guidelines might be developed as to which candidates should be followed up first and further studied with comprehensive functional validation using animal models and human cell lines that reproduce the BCA breakpoints.
Overall our results suggest that the integration of phenomatch scores, altered chromatin contacts, and other clinical gene annotations provide valuable interpretation to many variants of uncertain significance through long-range position effects. The correct prediction of 52 out of 57 known pathogenic genes in DGAP cases used as positive controls supports such integration. Our computational analysis is rapid and can provide additional information to benefit the clinical assessment of both coding and non-coding genome variants. The latter is an important step towards prediction of pathogenic consequences of non-coding variantion observed in prenatal samples. For example, based on its position and chromatin contact alterations, we correctly predicted the involvement and decreased expression of SOX9 in the cleft palate Pierre-Robin sequence (PRBNS [MIM: 261800]) association in DGAP288.(Ordulu et al. 2016)
Lastly, we would like to note that predicting the pathogenic outcome of disrupted chromatin contacts is not a straightforward endeavor: it has been shown that a single gene promoter can be targeted by several enhancers,(Thurman et al. 2012) therefore compensating for the perturbed interactions by the chromosome rearrangements. In addition, rearrangements can reposition gene promoters and enhancers outside of their preferred chromatin environments, leading to improper gene activation by enhancer adoption.(Lupiáñez et al. 2015) Our method currently identifies instances in which known and predicted enhancer/promoter interactions are disrupted by the rearrangement breakpoints and thus lead to decreased candidate gene expression. Enhancer adoption prediction will be incorporated once mathematical models of TAD formation upon changes in genomic sequence are refined and available to the greater scientific community. Presently, our predictions are as good as the availability of pathogenic gene annotations, chromatin conformation data, clinical phenotype information, and the presence of similar rearrangements in databases such as DECIPHER and dbVar. While the existence of other subjects with related phenotypes to the DGAP cases does not prove the involvement of neighboring genes in the etiology of these phenotypes, it is a step forward towards prediction of pathogenic effects starting from a simple computational analysis, pointing to a better phenotypic categorization when clinically examining affected individuals. By making our position effect prediction method available to the human genetics community, we hope to study additional cases with complete phenotypic information and be able to refine better the rules for the prediction of position effects on gene expression and discover new mechanisms of pathogenicity.